Depth of Sequencing Coverage
First, lets start with a few defintions:
-
Sequencing coverage refers to the percent (or total number) of target bases that have been sequenced relative to a reference set of bases. In whole-exome sequencing different platforms, such as illumina and agilent, have different bait sets that sequence different regions of the genome and therefore will have different references. Thus, providing a reference set of sequenced regions will not only speed up computational methods for determining sequencing depth, but it will also enable acurate quantification of sequencing depth. Whole-genome sequencing, by definition, sequences all bases included in the genome.
-
Sequencing depth refers to how many times each coverage base is sequenced on average. Sequencing depth is an important consideration to any next-generation sequencing (NGS) analysis, because it plays a critical role in our ability to call germline and somatic mutations. The higher (deeper) the sequencing depth, the more statistically powered we are to identify true alterations and differentiate them from artifacts (i.e. false positives). For example, when calling germline alterations we expect the alterations to have a variant allele fraction of 50% (i.e. alteration at locus on 1 copy of chromosome). One way we can quickly determine if the germline alteration call is a false positive is via a binomial distribution where the expected probability of success is 0.5 (e.g.
binom.test(alt_reads, depth, p=0.5)).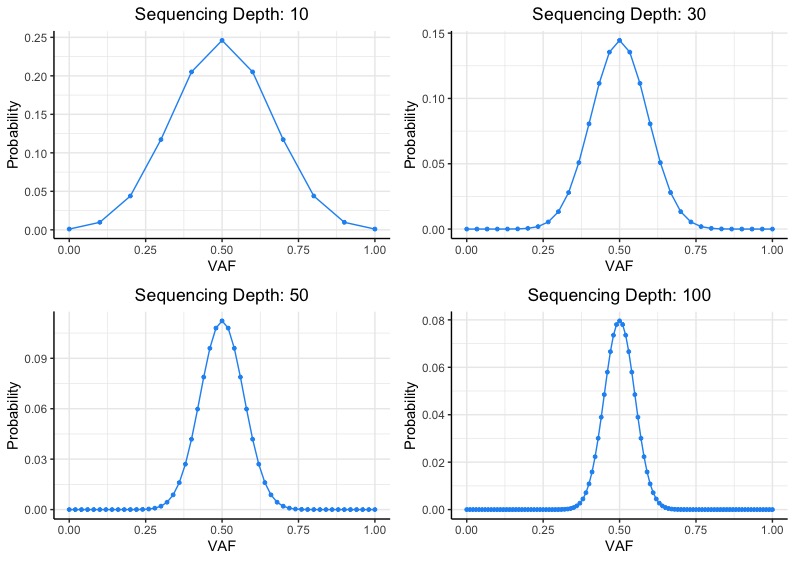 If the probability that the proportion of alternate reads given the sequencing depth significantly deviates from the expected VAF of a germline alteration (50%), then the germline alteration call is likely an artifact. As we can see from the plots above, the higher the sequencing depth, the more confident we are that observed VAF for a true germline alteration call will be closer to 50%. Conversely, looking at the top left panel (sequencing depth of 10), we cannot confidently say that a germline alteration with an observed VAF of 30% significantly deviates from the expected VAF of 50%. In practice, one should also look at IGV snapshots of the germline alterations when performing artifact filtering (if feasible).
If the probability that the proportion of alternate reads given the sequencing depth significantly deviates from the expected VAF of a germline alteration (50%), then the germline alteration call is likely an artifact. As we can see from the plots above, the higher the sequencing depth, the more confident we are that observed VAF for a true germline alteration call will be closer to 50%. Conversely, looking at the top left panel (sequencing depth of 10), we cannot confidently say that a germline alteration with an observed VAF of 30% significantly deviates from the expected VAF of 50%. In practice, one should also look at IGV snapshots of the germline alterations when performing artifact filtering (if feasible).For somatic mutation calling, sequencing depth primarily affects our ability to call subclonal mutations (i.e. mutations that are only in a fraction of tumor cells). The lower the cancer cell fraction (CCF), the higher the sequence coverage required to confidently call the mutations. For example, MuTect requires at least 5 supporting alternate reads to confidently call somatic mutations. The ABSOLUTE paper provides an excellent explanation on how tumor purity, local copy number, and sequencing depth affect the power to call mutations.
DepthOfCoverage (GATK3.7)
To run DepthOfCoverage, we can run the following command line code:
java -Xmx15g -jar /usr/GenomeAnalysisTK.jar \
-R ${refFasta} \
-T DepthOfCoverage \
-o ${sampleName} \
-omitBaseOutput \
-geneList ${geneList} \
-I $bamFile \
-L ${intervalList} \
--minBaseQuality ${minBaseQuality} \
--minMappingQuality ${minMappingQuality}
The descriptions of the inputs are as follows:
-R Reference FASTA file for the genome reference used (e.g. Hg19 or Hg38)
-o What suffix to append to the output files. Here we recommend the sample name/identifier
-geneList A refSeq file containing gene regions for coverage metrics to be aggregated over (provide if you want gene-level coverage metrics in addition to sample level coverage metrics)
-I BAM file that we want coverage information on
-L The genomic intervals (regions) that we want coverage information on. For whole-exomes these would be the regions captured by the bait kits. For genomes this can be left empty.
–minBaseQuality The minimum base quality, determined by the sequencer, to be counted in the coverage metrics
–minMappingQuality The minimum mapping quality, determined by the alignment algorithm (e.g. BWA), to be counted in the coverage metrics